
218 slide Skeletal Bone Genetics MARFAN SYNDROME PowerPoint Presentation on CD For Sale

When you click on links to various merchants on this site and make a purchase, this can result in this site earning a commission. Affiliate programs and affiliations include, but are not limited to, the eBay Partner Network.

218 slide Skeletal Bone Genetics MARFAN SYNDROME PowerPoint Presentation on CD :
$12.99
Thank you!
If you do not wish to have your item(s) delivered on data disc(s), I can provide them on a flash drive and other means as well. Just let me know if a disc does not work for you and we can discuss delivery by other methods.
COMBINING SHIPPING COSTS
Are you purchasing multiple items? I will: a) combine all invoices before payment and charge shipping equivalent to one item, or b) refund all shipping costs in excess of one item after payment.
All derivative (i.e. change in media; by compilation) work from this underlying U.S. Government public domain/public release data is COPYRIGHT © GOVPUBS$3.00 first class shipping in U.S. and rest of world.
Includes the Adobe Acrobat Reader for reading and printing publications.
Numerous illustrations and matrices.
Contains the following key public domain (not copyrighted) U.S. Government publication(s) on one CD-ROM in both Microsoft PowerPoint and Adobe Acrobat PDF file formats:
TITLE:
Marfan Syndrome, 218 pages (slides)
SLIDE TOPICS, SUBTOPICS and CONTENTS:Marfan Syndrome
Hang T. Nguyen, CPT, MC
Rheumatology Service
WRAMC
History
The Marfan syndrome (MFS) was first described in 1896 by a French pediatrician, Antoine Marfan
He documented the clinical signs of a 5 ½ year old girl with long thin limbs
Epidemiology
One of the most common inherited disorders of connective tissue
Autosomal dominant inheritance, variable penetrance
Incidence 1 in 10,000 to 20,000 individuals
Equal gender and ethnic distribution
Question
Mutations on which gene are associated with Marfan Syndrome?
Genetics
FBN1
Elastic and nonelastic tissue
Chromosome 15q21.1
More than 97 mutations identified
No correlation between specific mutation and clinical phenotype
25% MFS cases result from spontaneous mutations
Bonus Question
What heritable disorder of connective tissue is associated with mutations in the Fibrillin-2 gene?
Skeletal Manifestations
Arachnodactyly
Cardiac Manifestations
Aortic disease is main cause of morofferity and mortality in MFS
Dilatation found in 50% of children
60 to 80% of adults have aortic root dilatation
Cardiac Manifestations
Aortic Disease
Cystic medial necrosis
Other cardiac disease
Mitral valve prolapse leading to mitral regurgitation
Seen in 60-80% of MFS by echocardiogram
More common in women
Age-dependent
Pregnancy
Pregnant women are at particular risk for aortic dissection
Complications are most often seen in the second and third trimester
C-section
ß-blockers
Ocular Manifestations
Ectopia lentis
50 to 80 percent of MFS patients
Usually bilateral
Lens upwardly displaced
Other Findings
Dural Ectasia (Major Manifestation)
Lumbosacral region
Occurs in >90% of MFS patients
MRI most sensitive for diagnosis
Clinical significance?
Other Manifestations
Pulmonary System
Spontaneous pneumothorax
Apical blebs
Integumentary System
Striae atrophicae
Recurrent or incisional hernia
Marfan Syndrome
Diagnosis of MFS and other related conditions are based on clinical features
Detection of a fibrillin mutation contributes to the diagnostic criteria, but the diagnosis is primarily clinical
Diagnostic Requirements for MFS
Index case
If family or genetic history is not contributory
Major criteria in at least two different organ systems
Involvement of a third organ system
If the mutation known to cause Marfan syndrome in others is detected
One major criterion in an organ system
Involvement of a second organ system
Diagnostic Requirements for MFS
Relative of Index case
Major criterion in an organ system
Involvement of a second organ system
Skeletal System
Major if at least 4; Involved if either 2 major or 1 major and 2 minor
Major
Pectus carinatum or pectus excavatum (requiring surgery)
(+) wrist & thumb signs
Scoliosis >20 ° or kyphosis
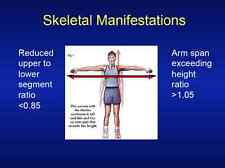
Reduced upper/lower segment (<0.85)
Arm span/height ratio >1.05
Reduced elbow extension
Protrusio acetabulae
Pes planus
Minor
Pectus excavatum (moderate)
Joint hypermobility
High arched palate with dental crowding
Typical facies, including
Dolichocephaly
Malar hypoplasia
Retrognathia
Enophthalmos
Downslanting palpebral fissures
Ocular system
Major if
Ectopia lentis
Involved if (two or more)
Flat cornea
Increased axial length of globe leading to myopia
Hypoplastic iris or ciliary muscle causing decreased miosis
Cardiovascular system
Major if (1 or more)
Dilatation of the ascending aorta
Dissection of the ascending aorta
Involved if (1 or more)
Mitral valve prolapse
Dilatation of pulmonary artery
Mitral annular calcification
Dilatation or dissection of descending aorta
Mitral regurgitation
Left ventricular dilatation
Tricuspid valve prolapse
Neurologic
Major
Lumbosacral dural ectasia (by CT or MRI)
Involved (if 1 or more)
Spontaneous pneumothorax
Apical blebs on CXR
Striae atrophicae
Recurrent or incisional hernia
Question
A 23 year-old man is referred to you for evaluation after having suffered a pathologic vertebral fracture. He is tall (6 ft 7 in), has dolichostenomelia, arachnodactyly and pectus excavatum and scoliosis as well as downward displacement of both lenses. No murmurs are audible on exam. He shows moderate mental retardation. His parents and siblings are of normal stature with no musculoskeletal abnormalities.
Does he have Marfan syndrome?
What would be the prime consideration in your differential given the clinical history?
Homocystinuria
Triad of mental retardation, connective tissue disorder and thrombosis
Differential Diagnoses of Marfan Syndrome
Homocystinuria
MASS phenotype
Congenital contractural arachnodactyly
Isolated ectopia lentis
Ehlers-Danlos syndromes types I, II and III
Stickler syndrome (hereditary arthroophthalmopathy)
Klinefelter syndrome
Question
A 23-year-old woman is self-referred for a routine examination. She has no specific complaints and has no symptoms on review of systems. Physical examination shows a tall, thin, young woman with a mild pectus excavatum, an increased arm span–to–height ratio, and arachnodactyly. Her pulse rate is 80/min and regular, respiratory rate is 16/min, and blood pressure is 120/60 mm Hg. Cardiac examination shows a loud S1 and a physiologically split S2 with no murmurs or gallops. An echocardiogram shows loss of the aortic sinotubular junction with mild aortic root dilatation (40 mm versus an expected dimension of 34 mm for her age and body size).
Which of the following is most appropriate?
Question
Provide reassurance; no further evaluation or therapy is needed.
Recommend echocardiography for all her first-degree relatives.
Initiate therapy with an angiotensin-converting enzyme inhibitor.
Discuss the need for permanent contraception.
Encourage increased physical activity.
Question
Provide reassurance; no further evaluation or therapy is needed.
Recommend echocardiography for all her first-degree relatives.
Initiate therapy with an angiotensin-converting enzyme inhibitor.
Discuss the need for permanent contraception.
Encourage increased physical activity.
Cardiac Causes of Sudden Death in Competitive Athletes
Life Expectancy in MFS
Management
Restriction of vigorous physical activity
Monitoring of aortic size
ß-blockers
Elective aortic root repair
Aortic root diameter > 55mm
50mm if considering pregnancy
Management
Annual ophthalmology exam
Semi-annual orthopedic evaluation
Bracing
Surgical intervention when scoliosis>40 degrees
Hormonal advancement of pubarche
Famous People with MFS
Take Home Points
Aortic dissection is the primary cause of death in MFS
Periodic echocardiographic evaluation is key in management of aortic dilatation
Echocardiographic screening for first-degree relatives
QUESTIONS?
Marfan Syndrome
LCDR Michael Keith
Rheumatology Fellow
March 23, 2005
Overview
History
Epidemiology
Genetics
Clinical features
Diagnosis
Mortality
Management
History
First described 1896
Dr. Antoine Marfan
Medical Society of Paris Hospitals
5 year-old female
Other Historical Figures
History
1914: Adoption of Marfan syndrome
Arachnodactyly
Dislocated lenses
1931: Autosomal dominant inheritance
1943: Association with aortic dissection
Epidemiology
Common inherited connective tissue disorder
Autosomal dominant, variable penetrance
Incidence about 1 in 10,000
Epidemiology
Equal gender and ethnic distribution
25% cases are sporadic
Arise from new mutation
Spectrum of Hereditary Disorders
Genetics
Late 1980s: Abnormalities of fibrillin
Important component of elastic tissues
Skin, aorta, arteries, ligaments, lung parenchyma
1991: Dietz et al.
Mutation of Fibrillin-1 gene
Mostly on chromosome 15
Over 50 mutations described
One family with mutation unknown gene chromosome 3
Genetics
Variable penetrance of fibrillin-1 mutation
Incomplete forms of disease
Milder or non-existant cardiovascular features
MASS
Myopia, MVP, Aorta, Skin, Skeletal
Isolated ectopia lentis
Clinical Features
Skeletal
Habitus
Hypermobility
Cardiovascular
Murmur
Echocardiographic abnormalities
Ocular
Lens displacement
Diagnosis
De Paepe et al. 1996
Clinical and Genetic Features
Ghent Nosology
Major criteria in 2 organ systems
Involvement of third organ system
Less stringent when + family hx
Diagnosis
Skeletal: Pectus
Excavatum
Carinatum
Skeletal: Dolichostenomelia
Skeletal: Steinberg’s sign
Positive when:
Thumb enclosed in the clenched fist
Nail extends beyond hypothenar border
Skeletal: Walker-Murdoch sign
Overlap of thumb and 5th digit as they encircle wrist
Skeletal: Arachnodactyly
Skeletal: Metacarpal index
Mean of the length divided by midpoint width of the 2nd – 4th metacarpals
Normal
5.4 to 7.9
MFS
>8.4
Skeletal: Spine involvement
Scoliosis > 20°
Kyphosis
Skeletal: Protrusio acetabulae
Positon of medial wall acetabulum c/w ilioischial line
Women
Normal: 1 mm medial
MFS: 6 mm medial
Men
Normal: 2 mm lateral
MFS: 3 mm medial
Skeletal: Miscellaneous
Major
Elbow extension <170°
Pes planus
Involvement
Minor pectus excavatum
Joint hypermobility
High arched palate
Typical facies
Dolichocephaly
Malar hypoplasia
Retrognathia
Enophthalmos
Downslanting palpebral fissures
Ocular: Ectopia Lentis
About 60% of cases
Typically bilateral
Upward displacement
Ocular: Involvement
Flat cornea
Increased axial length of globe
Myopia
Hypoplastic iris or ciliary muscle
Decreased miosis
Cardiovascular: Major
Dilatation of Aortic Root
Includes sinus of Valsalva
Dissection of Ascending Aorta
Stanford A
DeBakey I,II
Cardiovascular: Involved
Mitral valve prolapse
PA dilatation, age < 40
Mitral Annular Calcification, age < 40
Other Aortic dilatation or dissection
Pulmonary
Major
None defined
Involvement
Spontaneous pneumothorax
Apical blebs
Cutaneous
Major (none)
Minor
Striae atrophicae
Recurrent hernia
Incisional hernia
Differential Diagnosis
Homocystinuria
Ectopia lentis (downward displacement)
Mental retardation
Risk of thrombosis
MASS phenotype
mitral valve prolapse
aortic dilatation
skin involvement
skeletal involvement
lower risk of aortic aneurysm rupture
Differential Diagnosis
Congenital contractural arachnodactyly
Mutation of fibrillin-2 gene, chromosome 5
80% homology to fibrillin-1
Marfanoid habitus, no cardiac or eye findings
Isolated ectopia lentis
Associated with fibrillin-1 gene
No other findings
Differential Diagnosis
Ehlers-Danlos syndromes (I,II,III)
Stickler syndrome
Hereditary arthro-ophthalmopathy
Type II collagen
Klinefelter syndrome
XXY genotype
Tall stature, small testes, gynecomastia
No cardiac or ocular manifestations
Mortality
Aortic dissection
Main cause of premature death
Improved life expectancy
1972 mean 32 years
1995 mean 45 years
Mortality
Decrease in death from aortic dissection
1972: 70%
1995: 48%
Improved medical and surgical management
Sudden Cardiac Death
Often excel in sports
Height: volleyball, basketball
Flexibility: ballet, gymnastics
Aortic Dissection in Marfan Syndrome
Study by Maron et al. JAMA 1996;276:199-204.
Third most common cause of SCD in athletes
Frequency = 5%
Management
Focus on cardiovascular complications
Serial echocardiogram
Baseline
Every 6-12 months
Management
Risk factors for aortic dissection
Aortic diameter > 5 cm
Dilatation beyond sinus of Valsalva
Family history
Rapid rate of increased dilatation
> 5 % / year
> 2 mm / year in adults
Management
Pregnancy
Increased risk of aortic dissection
Gestational hypertension
Pre-eclampsia
Especially when aorta > 4 cm
Frequent monitoring during pregnancy and puerperium
Medical Therapies
β-blockers (usually lifelong)
Atenolol, metoprolol, propranolol
Decrease aortic stiffness and pulse velocity
Most effective in aorta < 4cm
? Calcium channel blockers
? Angiotensin inhibition (Ace-I / Arb)
Surgical Therapy
Better outcome with early surgery
Worse outcome with late or emergent surgery
Referral for elective surgery
Adults with aorta > 5.5 cm
Children with aorta > 5.0 cm
Other System Management
Annual ophthalmology exam
Attempts of refractive correction
Surgery may be required for ectopia lentis
Semi-annual orthopedic evaluation
Bracing
Surgical intervention when scoliosis>40 degrees
Conclusion: Key Points
Diagnosis is largely clinical
Aortic dissection = primary cause of death
Remember SCD in athletes
Periodic echocardiographic evaluation
Screening for 1st-degree relatives
Consider lifelong beta-blockade
Genetic counselling
Questions?
Marfan Syndrome
LCDR Michael Keith
WRAMC Rheumatology
January 10, 2006
Overview
History
Epidemiology
Genetics
Diagnosis and Clinical features
Mortality
Management
Recognition of Features
1914: Adoption of Marfan syndrome
Arachnodactyly
Dislocated lenses
1931: Autosomal dominant inheritance
1943: Association with aortic dissection
Epidemiology
Common inherited connective tissue disorder
Autosomal dominant, variable penetrance
Incidence about 1 in 10,000
Epidemiology
Equal gender and ethnic distribution
25% cases are sporadic
Arise from new genetic mutation
Genetics
Late 1980s: Abnormalities of fibrillin
Important component of elastic tissues
Skin, aorta, arteries, ligaments, lung parenchyma
1991: Dietz et al.
Mutation of Fibrillin-1 gene
Mostly on chromosome 15
More than 100 mutations described
One family described
Mutation of unknown gene chromosome 3
Fibrillin
Important protein
Elastic connective tissues
Also non-elastic tissues
At least 2 forms
Fibrillin-1
Fibrillin-2
Genetics
Variable penetrance of fibrillin-1 mutation
Incomplete forms of disease
Milder or non-existant cardiovascular features
MASS
Myopia, MVP, Aorta, Skin, Skeletal
Isolated ectopia lentis
Key Clinical Features
Skeletal
Habitus
Hypermobility
Cardiovascular
Murmur
Echocardiographic abnormalities
Ocular
Lens displacement
Diagnosis
De Paepe et al. 1996
Clinical and Genetic Features
Ghent Nosology
Major criteria in 2 organ systems
Involvement of third organ system
Less stringent when + family hx
Ghent Nosology
Skeletal System
Major if at least 4; Involved if either 2 major or 1 major and 2 minor
Major
pectus carinatum
pectus excavatum (requiring surgery)
(+) wrist & thumb signs
scoliosis >20 ° or spondylolithesis
reduced upper/lower segment (>0.85) or arm span/height ratio >1.05
reduced elbow extension
pes planus
Minor
pectus excavatum (moderate)
joint hypermobility
high arched palate with dental crowding
typical facies, including
Dolichocephaly
malar hypoplasia
retrognathia
enophthalmos
downslanting palpebral fissures
Steinberg’s sign
Thumb enclosed in the clenched fist
Nail extends beyond hypothenar border
Walker-Murdoch Sign
Overlap of thumb and 5th digit as they encircle wrist
Skeletal: Arachnodactyly
Metatarsal Index
Recently proposed in Marfan syndrome
Not validated in other papers
Measurements similar to MCI
Average metatarsal index
12.7 in Marfan patients
9.8 in controls (P<0.0005)
Spine Involvement
Scoliosis > 20°
Kyphosis
Protrusio Acetabulae
Positon of medial wall acetabulum c/w ilioischial line
Women
Normal: 1 mm medial
MFS: 6 mm medial
Men
Normal: 2 mm lateral
MFS: 3 mm medial
Ocular System
Major
Ectopia lentis
Involved if (two or more)
Flat cornea
Increase axial length of globe
Hypoplastic iris or ciliary muscle causing decreased miosis
Ectopia Lentis
About 60% of cases
Typically bilateral
Upward displacement
Cardiovascular System
Major if (1 or more)
dilation of the ascending aorta
dissection of the ascending aorta
Involved if (1 or more)
MVP
dilation of pulmonary artery <40 years
MACC <40 years
dilation or dissection of descending aorta <50 years
Mitral regurgitation
Left ventricular dilatation
Tricuspid valve prolapse
Aortic Disease
Cystic medial necrosis
Aortic Root Aneurysm
Aortic Root Dilatation
Pulmonary
Major
None defined
Involvement
Spontaneous pneumothorax
Apical blebs
Differential Diagnosis
Homocystinuria
Ectopia lentis (downward displacement)
Mental retardation
Risk of thrombosis
MASS phenotype
mitral valve prolapse
aortic dilatation
skin involvement
skeletal involvement
lower risk of aortic aneurysm rupture
Differential Diagnosis
Congenital contractural arachnodactyly
Mutation of fibrillin-2 gene, chromosome 5
80% homology to fibrillin-1
Marfanoid habitus, no cardiac or eye findings
Isolated ectopia lentis
Associated with fibrillin-1 gene
No other findings
Differential Diagnosis
Ehlers-Danlos syndromes (I,II,III)
Stickler syndrome
Hereditary arthro-ophthalmopathy
Type II collagen
Klinefelter syndrome
XXY genotype
Tall stature, small testes, gynecomastia
No cardiac or ocular manifestations
Question
A 23-year-old woman is self-referred for a routine examination. She has no specific complaints and has no symptoms on review of systems. Physical examination shows a tall, thin, young woman with a mild pectus excavatum, an increased arm span–to–height ratio, and arachnodactyly. Her pulse rate is 80/min and regular, respiratory rate is 16/min, and blood pressure is 120/60 mm Hg. Cardiac examination shows a loud S1 and a physiologically split S2 with no murmurs or gallops. An echocardiogram shows loss of the aortic sinotubular junction with mild aortic root dilatation (40 mm versus an expected dimension of 34 mm for her age and body size).
Which of the following is most appropriate?
Question
(A) Provide reassurance; no further evaluation or therapy is needed.
(B) Recommend echocardiography for all her first-degree relatives.
(C) Initiate therapy with an angiotensin-converting enzyme inhibitor.
(D) Discuss the need for permanent contraception.
(E) Encourage increased physical activity.
Question
(A) Provide reassurance; no further evaluation or therapy is needed.
(B) Recommend echocardiography for all her first-degree relatives.
(C) Initiate therapy with an angiotensin-converting enzyme inhibitor.
(D) Discuss the need for permanent contraception.
(E) Encourage increased physical activity.
Mortality
Aortic dissection
Main cause of premature death
Aortic dilatation in 50% of children with MFS
Improving life expectancy
1972 mean 32 years
1995 mean 45 years
Life Expectancy In MFS
Mortality
Decrease in death from aortic dissection
1972: 70%
1995: 48%
Improved medical and surgical management
Sudden Cardiac Death
Marfan Patients often excel in sports
Height: volleyball, basketball
Flexibility: ballet, gymnastics
Aortic Dissection in Marfan Syndrome
Maron et al. JAMA 1996;276:199-204.
Third most common cause of SCD in athletes
Frequency = 5%
Cardiac Causes of Sudden Death in Competitive Athletes
Management
Awareness of risk factors for aortic complications
Focus on cardiovascular complications
Medical
Surgical
Serial echocardiogram
Baseline
Every 6-12 months
Management
Risk factors for aortic dissection
Aortic diameter > 5 cm
Dilatation beyond sinus of Valsalva
Family history
Rapid rate of increased dilatation
> 5 % / year
> 2 mm / year in adults
Management
Pregnancy
Increased risk of aortic dissection
Gestational hypertension
Pre-eclampsia
Especially when aorta > 4 cm
Frequent monitoring during pregnancy and puerperium
Medical Therapies
β-blockers (usually lifelong)
Atenolol, metoprolol, propranolol
Decrease aortic stiffness and pulse velocity
Most effective in aorta < 4cm
? Calcium channel blockers
? Angiotensin blockade
Recent data suggest benefit for Enalapril
Yetman AT, Bornemeier RA, McCrindle BW. Am J Cardiol. 2005 May 1;95(9):1125-7.
Surgical Therapy
Better outcome with early surgery
Worse outcome with late or emergent surgery
Referral for elective surgery
Adults with aorta > 5.5 cm
Children with aorta > 5.0 cm
Other System Management
Annual ophthalmology exam
Attempts of refractive correction
Surgery may be required for ectopia lentis
Semi-annual orthopedic evaluation
Bracing
Surgical intervention when scoliosis>40 degrees
Key Points
Diagnosis is largely clinical
Aortic dissection = primary cause of death
Remember SCD in athletes
Periodic echocardiographic evaluation
Screening for 1st-degree relatives
Consider lifelong beta-blockade
Genetic counseling
Questions?
Marfan Syndrome
Michael A. Malloy, M.D., M.P.H.
Rheumatology Service
WRAMC
History
The Marfan syndrome (MFS) was first described in 1896 by a French pediatrician, Antoine Marfan
He documented the clinical signs of a 5 ½ year old girl with long thin limbs
Epidemiology
MFS is one of the most common inherited disorders of connective tissue
Autosomal dominant inheritance
Incidence 1 in 10,000 to 20,000 individuals
Pregnancy
Pregnant women are at particular risk for aortic dissection
Complications are most often seen in the second and third trimester
C-section
ß-blockers
Musculoskeletal manifestations
Arachnodactyly
Dolichostenomelia
Other findings
Ectopia lentis
Dural ectasia
Integument
Marfan Syndrome
Diagnosis of MFS and other related conditions are based on clinical features
Mutations in the FBN1, the gene that encodes fibrillin-1, are responsible for MFS
Diagnostic Requirements for MFS
Index case
If family or genetic history is not contributory
Major criteria in at least two different organ systems
Involvement of a third organ system
If the mutation known to cause Marfan syndrome in others is detected
One major criterion in an organ system
Involvement of a second organ system
Diagnostic Requirements for MFS
Relative of Index case
Presence of a major criterion in the family history
Major criterion in an organ system
Involvment of a second organ system
Skeletal System
Major if at least 4; Involved if either 2 major or 1 major and 2 minor
Major
pectus carinatum
pectus excavatum (requiring surgery)
(+) wrist & thumb signs
scoliosis >20 ° or spondylolithesis
reduced upper/lower segment (>0.85) or arm span/height ratio >1.05
reduced elbow extension
pes planus
Minor
pectus excavatum (moderate)
joint hypermobility
high arched palate with dental crowding
typical facies, including
Dolichocephaly
malar hypoplasia
retrognathia
enophthalmos
downslanting palpebral fissures
Ocular system
Major if
Ectopia lentis
Involved if (two or more)
Flat cornea
Increase axial length of globe
Hypoplastic iris or ciliary muscle causing decreased miosis
Cardiovascular system
Major if (1 or more)
dilation of the ascending aorta
dissection of the ascending aorta
Involved if (1 or more)
MVP
dilation of pulmonary artery <40 years
MACC <40 years
dilation or dissection of descending aorta <50 years
Mitral regurgitation
Left ventricular dilatation
Tricuspid valve prolapse
Skin & Integument
Major
lumbosacral dural ectasia (by CT or MRI)
Involved (if either)
striae atrophicae
recurrent or incisional hernia
Pulmonary
Involved (if either)
spontaneous pneumothorax
apical blebs on CXR
Differential Dx of Marfan syndrome
Homocystinuria
MASS phenotype
Congenital contractural arachnodactyly
Isolated ectopia lentis
Ehlers-Danlos syndromes types II and III
Stickler syndrome (hereditary arthroophthalmopathy)
Klinefelter syndrome
Question
A 23-year-old woman is self-referred for a routine examination. She has no specific complaints and has no symptoms on review of systems. Physical examination shows a tall, thin, young woman with a mild pectus excavatum, an increased arm span–to–height ratio, and arachnodactyly. Her pulse rate is 80/min and regular, respiratory rate is 16/min, and blood pressure is 120/60 mm Hg. Cardiac examination shows a loud S1 and a physiologically split S2 with no murmurs or gallops. An echocardiogram shows loss of the aortic sinotubular junction with mild aortic root dilatation (40 mm versus an expected dimension of 34 mm for her age and body size).
Which of the following is most appropriate?
Question
(A) Provide reassurance; no further evaluation or therapy is needed.
(B) Recommend echocardiography for all her first-degree relatives.
(C) Initiate therapy with an angiotensin-converting enzyme inhibitor.
(D) Discuss the need for permanent contraception.
(E) Encourage increased physical activity.
Question
(A) Provide reassurance; no further evaluation or therapy is needed.
(B) Recommend echocardiography for all her first-degree relatives.
(C) Initiate therapy with an angiotensin-converting enzyme inhibitor.
(D) Discuss the need for permanent contraception.
(E) Encourage increased physical activity.
Cardiac Causes of Sudden Death in Competitive Athletes
Treatment
Life span
ß-blockers
Elective aortic root repair
Life expectancy in MFS
Treatment
Life span
ß-blockers
Elective aortic root repair
Question
All of the following conditions preclude participating in high-intensity competitive sports EXCEPT:
(A) Pulmonary hypertension of whatever cause
(B) Hypertrophic cardiomyopathy
(C) Ventricular septal defect with a pulmonic systemic blood flow ratio of less than 2
(D) Marfan’s syndrome with aortic root dilatation
(E) Symptomatic paroxysmal supraventricular tachycardia
Question
All of the following conditions preclude participating in high-intensity competitive sports EXCEPT:
(A) Pulmonary hypertension of whatever cause
(B) Hypertrophic cardiomyopathy
(C) Ventricular septal defect with a pulmonic systemic blood flow ratio of less than 2
(D) Marfan’s syndrome with aortic root dilatation
(E) Symptomatic paroxysmal supraventricular tachycardia
Famous people with MFS
Case
A 32-year-old woman with Marfan syndrome has mild dilatation of the ascending aorta with a maximum root dimension 1.3 times expected for her age and body size. She begins ß-blocker therapy, and both her children and her siblings undergo echocardiographic examinations. Annual echocardiographic examinations of the patient show a gradual increase from 42 to 48 mm in maximum root dimension. Four years after the initial diagnosis, the patient has a sudden onset of severe chest pain that radiates to her back.
Echocardiography shows acute ascending dissection, and the patient undergoes emergency aortic valve and root replacement.
Take Home Points
Aortic dissection is the primary cause of death among patients with Marfan syndrome
Periodic echocardiographic evaluation of aortic root dilatation is key in the treatment of patients with Marfan syndrome
First-degree relatives should undergo echocardiographic screening for this autosomal dominant disorder
Name that tune…
Marfan Syndrome (MFS)
Thomas Kremenski
Rheumatology Service
WRAMC
Heritable Collagen Diseases
Over 200 different heritable disorders of connective tissue (examples):
- Marfan Syndrome
- Homocystinuria
- Ehlers-Danlos Syndrome(s)
- Congenital Contractural Arachnodactyly
- Stickler Syndrome
Epidemiology of MFS
Prevalence = 1 case/3000-5000 in the U.S.
Approximately 50,000 cases in the U.S.
Both sexes affected equally
All races affected equally
Marfan Syndrome (MFS)
Major organ systems affected in MFS:
- Skeletal
- Ocular
- Cardiovascular
- Pulmonary
- Integumentary
- Nervous
- Neurological
History of MFS
First described by Dr. Antoine Marfan in 1896
1914: ectopia lentis
1931: autosomal dominant inheritance
1943: aortic dilatation and dissection
1970: ECHO introduced for MFS
1975: MVP
1988: dural ectasia
History of MFS cont.
1931: thought to be caused by defect in mesoderm
1955: MFS included in group of heritable D/Os
1986: clinical criteria for diagnosis in Berlin Nosology
1991: discovery of gene (15q21) on Chromosome 15 where mutation occurs in MFS
Famous people with MFS!
Cause of MFS
Mutation in gene 15q21 on Chromosome 15 which codes for the microfibrillar protein Fibrillin-1 (FBN1)
To date, more than 4 dozen mutations in the FBN-1 gene have been found in people with MFS
FBN1 is the major protein of connective tissue microfibrils essential to normal “elastic” fibrillogenesis
Cause of MFS cont.
Variable expression: defective gene expresses itself in different ways in different people (even with same gene mutation)
MFS is “autosomal dominant”: 50-50 chance of inheriting disorder if parent has MFS
Approximately 25% of cases occur as a result of a spontaneous mutation of the FBN-1 gene
Characteristics of MFS
(Skeletal System)
Patients are typically tall, slender, and loose-jointed
Dolichostenomelia: arm span > height by > 5%
Abnormally low US/LS ratio
- Measure total height and top of pubis symphysis to floor
- Normal white adult = 0.92
- Normal African-American = 0.87
Arachnodactyly: elongated fingers/toes
- also seen in other disorders (e.g., EDS, Homocystinuria)
- Positive “Thumb” and “Wrist” signs
- Abnormal “metacarpal index” done via radiograph
Characteristics of MFS cont.
(Skeletal System)
Pectus excavatum and carinatum
Loose-jointedness
- pes planus (flat feet)
- hyperextensibility of elbows, fingers, knees, hips
- recurrent dislocations of patella due to ligament laxity
Scoliosis, thoracic kyphosis, spondylolisthesis
Dolichocephaly: long and narrow face
Protusio acetabulae with deep hip socket
Congenital contractures of appendicular joints
Highly arched palate and crowding of teeth
Characteristics of MFS cont.
(Ocular System)
Most have myopia
More than 50% experience ectopia lentis: subluxation of lens (usually displaced upwards)
Retinal detachment
Glaucoma
Cataracts
Characteristics of MFS cont.
(Cardiovascular System)
At least 90% will have cardiovascular involvement
Dilatation of ascending aorta with or without regurgitation and involving at least sinuses of Valsalva
- Aortic dissection/rupture is the main cause of death (90%)
MVP occurs in approximately 80% and leads to significant mitral regurgitation in 10%
Characteristics of MFS cont.
(Integumentary System)
Increased striae (stretch marks) seen: poses no health risk
- over pectoral, deltoid, and lumbar areas
- helpful diagnostic sign
Increased risk of abdominal/inguinal hernias
Characteristics of MFS cont.
(Nervous System)
Dural ectasia: results from enlargement of spinal canal and can produce erosion of lumbar and sacral vertebrae
- requires MRI or CT to diagnose
- may lead to nerve root problems (radiculopathy)
Characteristics of MFS cont.
(Pulmonary System)
Generally not a common or significant problem
Apical bullae can lead to pneumothorax in 5%
Thoracic kyphosis associated with decreased lung capacity
Differential Diagnosis
Homocystinuria: diagnosis made by measurement of plasma amino acid concentration (e.g., total homocysteine)
Congenital Contractural Arachnodactyly: multiple contractures, arachnodactyly, and ext. ear malformations
MASS: myopia, MVP, mild aortic dilatation, skin and skeletal
Stickler Syndrome: myopia, vitreal degeneration, sensorineural hearing loss, hyper- or hypo-mobile joints, mandibular hypoplasia
Benign Hypermobility Syndrome: diagnose by Beighton’s citeria and exclude MFS, EDS, etc.
Diagnosis of MFS
Revised Diagnostic Criteria from 1996
- First published in American Journal of Medical Genetics
Relies on recognition of both “major”and “minor” clinical manifestations involving skeletal, cardiovascular, ocular pulmonary, and integumentary systems and dura
In general, there are “major” and “minor” criteria for each system (except pulmonary and skin - “minor” only)
“System Involvement”: additional criteria carrying less weight than “major” criterion
Diagnosis of MFS cont.
Family/Genetic History of MFS considered “major” criterian
“Major” include:
- 4 of 8 typical skeletal manifestations
- ectopia lentis
- aortic root dilatation involving sinuses of Valsalva
- LS dural ectasia by CT or MRI
- Family or genetic history of MFS
Diagnosis of MFS cont.
If patient has family history of MFS, you need:
- 1 “major” criterion in an organ system and “involvement” in a second organ system
If considered an “index” case, you need:
- “major” criteria from at least two systems and “involvement” of a third organ system
Diagnosis of MFS cont.
In general, the work-up will include:
- Cardiology consult and ECHO to assess for MVP, MR, aortic root dilatation, and AR
- Transesophageal ECHO, chest CT/MRI, aortography for suspected aortic dissection
- CXR
- Ophthalmological exam with slit lamp evaluation
- Orthopedic evaluation (e.g., scoliosis)
Management of MFS
Regular cardiac evaluation and annual ECHOs (q6 months
when aortic root diameter > 4.5 cm)
Endocarditis prophylaxsis
Use of beta- blokers to reduce progression of aortic root dilatation (all patients including children)
Elective aortic root replacement when aortic root diameter in the 5.0-6.0 cm range
Regular ophthalmological evaluation and treatment
Management of MFS cont.
Regular orthopedic evaluation and treatment (especially in growing children)
- back-bracing when spine curve in 20 - 40 degree range and surgery when 40-50 degree range
- surgery for severe pectus excavatum
- orthotics for flat feet
In pregnancy:
- need close regular follow-up by specialists experienced with MFS and counseling on risk of aortic dissection
Management of MFS cont.
Consider genetic counseling
Patient education regarding physical limitations
- should not engage in very strenuous activities such as weight lifting, wrestling, football, basketball, soccer, hockey, moving heavy furniture, or other activities that increase stress on aorta
- may engage in relatively non-strenuous, non-contact activities like golf, swimming, non,competitive biking
Address other psychosocial issues like self-esteem
- e.g., counseling and support groups
Prognosis of MFS
The average life span of untreated MFS was about 32 years in 1972
Early detection and medical and surgical treatment have increased the average life span dramatically by more than 25% from 48 years in 1972 to 72 years in 1996
INTRACITY GRAND ROUND
Walter M. Downs
Walter Reed Army Medical Center
15 February 2001
Case: Tall Stature
22 year-old male referred for evaluation of tall stature & left breast enlargement
He has no complaints
Differential Diagnosis
Marfan syndrome
Congenital contractural arachnodactyly
MASS phenotype
Ehlers-Danlos syndrome
Stickler syndrome
Homocystinuria
Endocrine disorder
Klinefelter syndrome
Fragile X syndrome
Acromegaly /Gigantism
History
22 year-old male
nickname of “tree” and “pretezel”
left breast / chest enlargement for 3 months
History
PMH/PSH
Left leg laceration
Social History
Tobacco 1/2 ppd
ETOH moderate
Active duty cook
History
Family History
Father 6’4”
Mother 5’11”
Sister 6’4”
No known inherited connective tissue disorder
Review of Systems
No joint dislocation, hernias or poor wound healing / scarring
No vision problems
No bleeding or easy bruisibility
Physical Exam
Gen: tall thin white male, height 6’9”
Heent: no high arched palate
Lung: CTA
CV: RRR w/o murmur or rub
Chest: bilateral gynecomastia, no other
deformity
Abd: (+) striae, no HSM or bruit
Physical Exam: Striae
No marked weight change
Not a weight lifter
No repetitive stress
Physical Exam
GU: small testes < 2 cm in size
Ext: no synovitis, FROM
no hyperextensible skin
scars with wide mouth
upper/lower ratio 0.8 (nl <0.85)
arm span/height ratio 1.0 (nl <1.05)
arachnodactyly w/ hyperextensible joints
pes planus
Physical Exam: Hypermobile Joints
Physical Exam: Arachnodactyly
Thumb (Steinberg) sign
is positive when the thumb,
enclosed in the clenched fist,
extends beyond the
hypothenar border
Physical Exam: Arachnodactyly
Wrist (Walker-Murdock) sign
is positive when there is
overlap of the thumb and
the 5th digit as they
encircle the opposite wrist
Ancillary Evaluation
DEXA Scan
osteoporotic
AP spine -2.89
hip - 1.03
Echocardiogram
normal study
Ophthalmology c/s
normal exam
Skeletal Survey
arachnodactyly
pes planus
no scoliosis
no spondylolithesis
no pectus deformity
Radiology: Arachnodactyly
Metacarpal index =
mean value of the length divided by the
mean mid point widths of
the 2nd, 3rd, & 4th metacarpals
Normal 5.4 - 7.9
Marfan > 8.4
Patient 9.75
Laboratory Evaluation
TSH 0.63 (0.5-4.9)
Homocystine 14 (8-20)
LH 19.7 (1.7-8.6)
FSH 55.4 (1.5-12.4)
Estradiol <20
Testosterone 114 (241-827)
Genetic Analysis 47 XXY
Diagnosis
KLINEFELTER SYNDROME
? Collagen defect
Marfan Syndrome
The Marfan syndrome (MFS), initially described just over 100 years ago, was among the first conditions classified as a heritable disorder of connective tissue.
MFS lies at one end of the phenotypic continuum, with people in the general population who have one or another feature of MFS at the other end.
Marfan Syndrome
Diagnosis of MFS and other related conditions remain based on clinical features.
Mutations in the FBN1, the gene that encodes fibrillin-1, are responsible for MFS.
In addition to skeletal, ocular and cardiovascular features, patients have involvement of the skin, integument, lungs and muscle tissue.
*Diagnostic Criteria for MFS
Major criteria in two systems,
Plus involvement of a third system
or
Family history positive
Major criteria in one organ system
Plus involvement of second system
From De Paepe et al., Am J Med Genet 62:417-426, 1996
Family History
First degree relative independently meets diagnostic criteria
Presence of a mutation in FBN1 know to cause Marfan syndrome
Presence of a haplotype around FBN1, inherited by descent, associated with unequivocally diagnosed Marfan syndrome in the family
Skeletal System
Major if at least 4; Involved if either 2 major or 1 major and 2 minor
Major
pectus carinatum
pectus excavatum (requiring surgery)
(+) wrist & thumb signs
scoliosis >20 or spondylolithesis
reduced upper/lower segment (>0.85) or arm span/height ratio >1.05
reduced elbow extension
pes planus
Minor
pectus excavatum (moderate)
joint hypermobility
high arched palate with dental crowding
typical facies, including
Dolichephaly
malar hypoplasia
retrognathia
enophthalmos
downslanting palpebral fiocssures
Ocular system
Major if
Ectopia lentis
Involved if (two or more)
Flat cornea
Increase axial length of globe
Hypoplastic iris or ciliary muscle causing decreased miosis
Cardiovascular system
Major if (1 or more)
dilation of the ascending aorta
dissection of the ascending aorta
Involved if (1 or more)
MVP
dilation of pulmonary artery <40 years
MACC <40 years
dilation or dissection of descending aorta <50 years
Skin & Integument
Major
lumbosacral dural ectasia (by CT or MRI)
Involved (if either)
striae atrophicae
recurrent or incisional hernia
Pulmonary
Involved (if either)
spontaneous pneumothorax
apical blebs on CXR
Questions?
Any further evaluation for collagen defect?
Recommended follow up?
Related Items:
218 slide Skeletal Bone Genetics MARFAN SYNDROME PowerPoint Presentation on CD
$12.99

T16 HISTORIC Glass Magic Lantern Slide WORLD PLACES 218 T ENAMI PALACE DISTANCE
$35.96

W60 HISTORIC Glass Magic Lantern Slide America 218 T ENAMI SNOW TRAIL TO WATER
$16.11